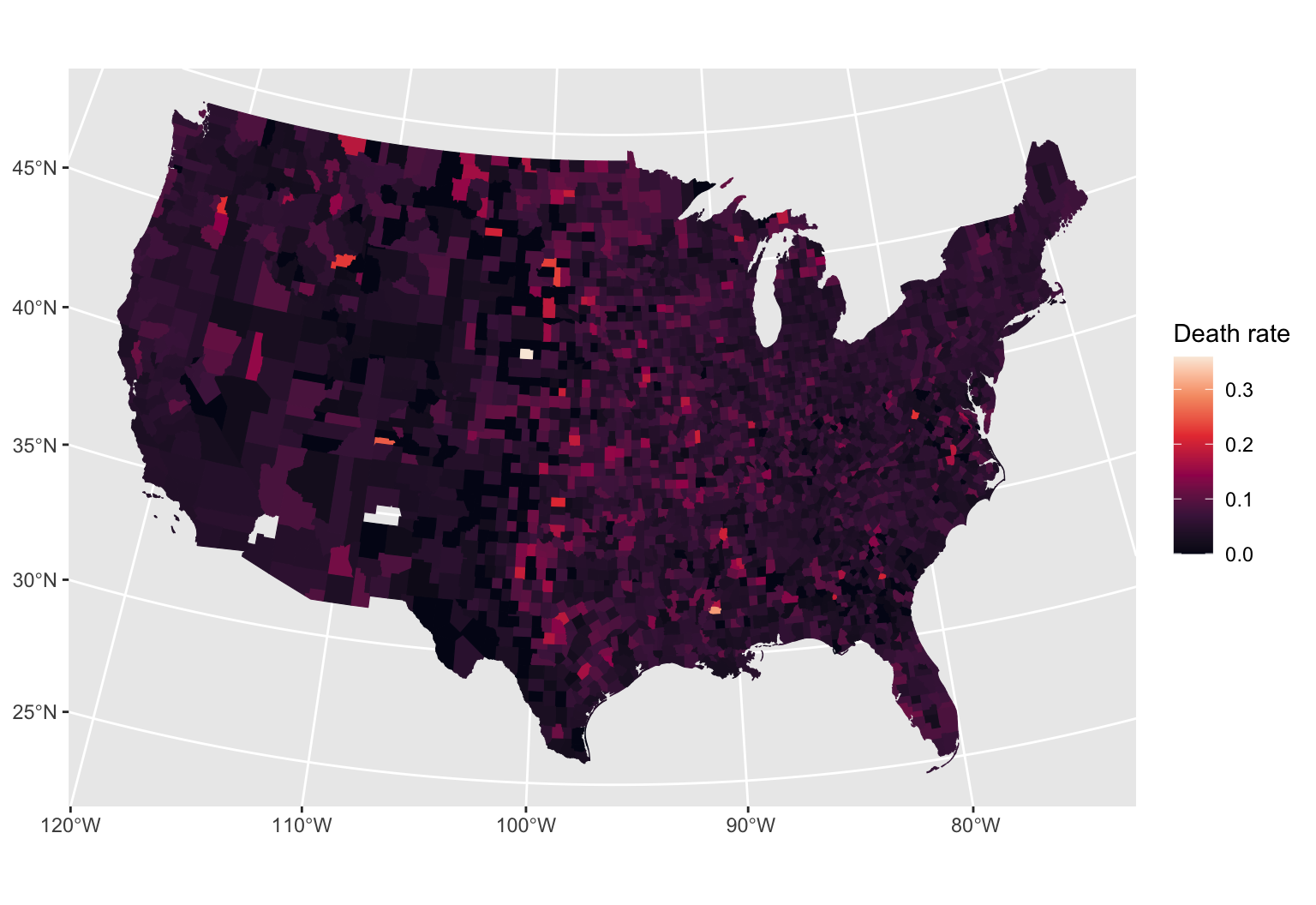
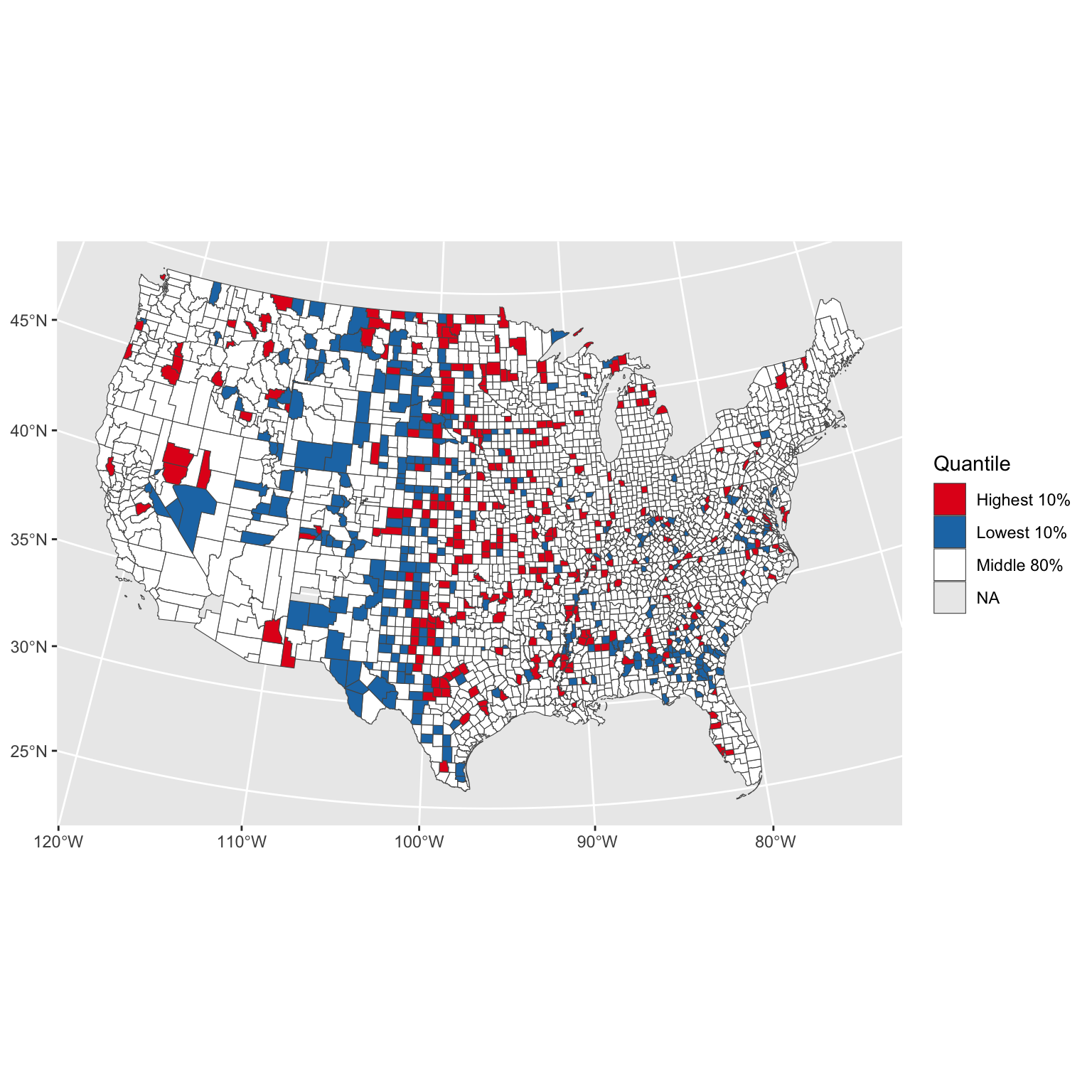
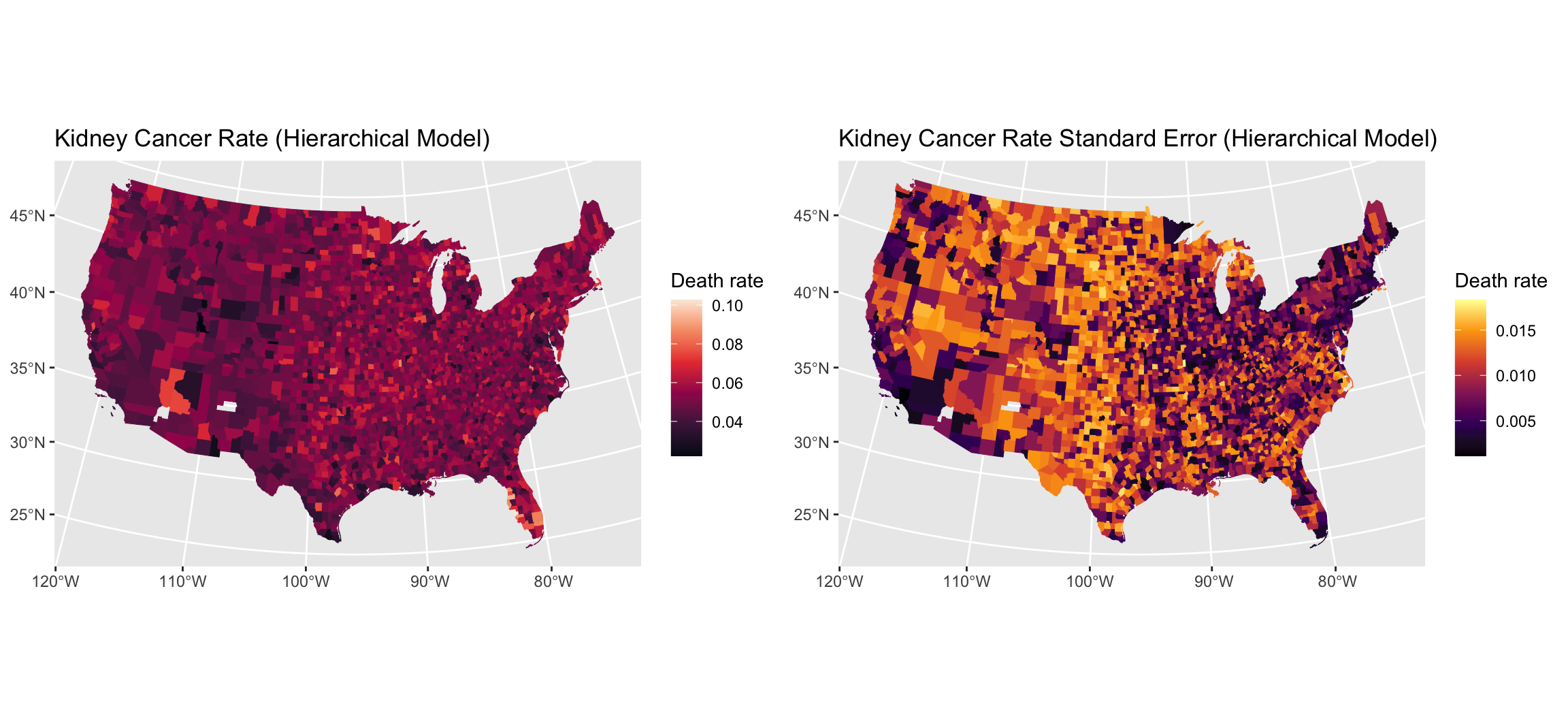
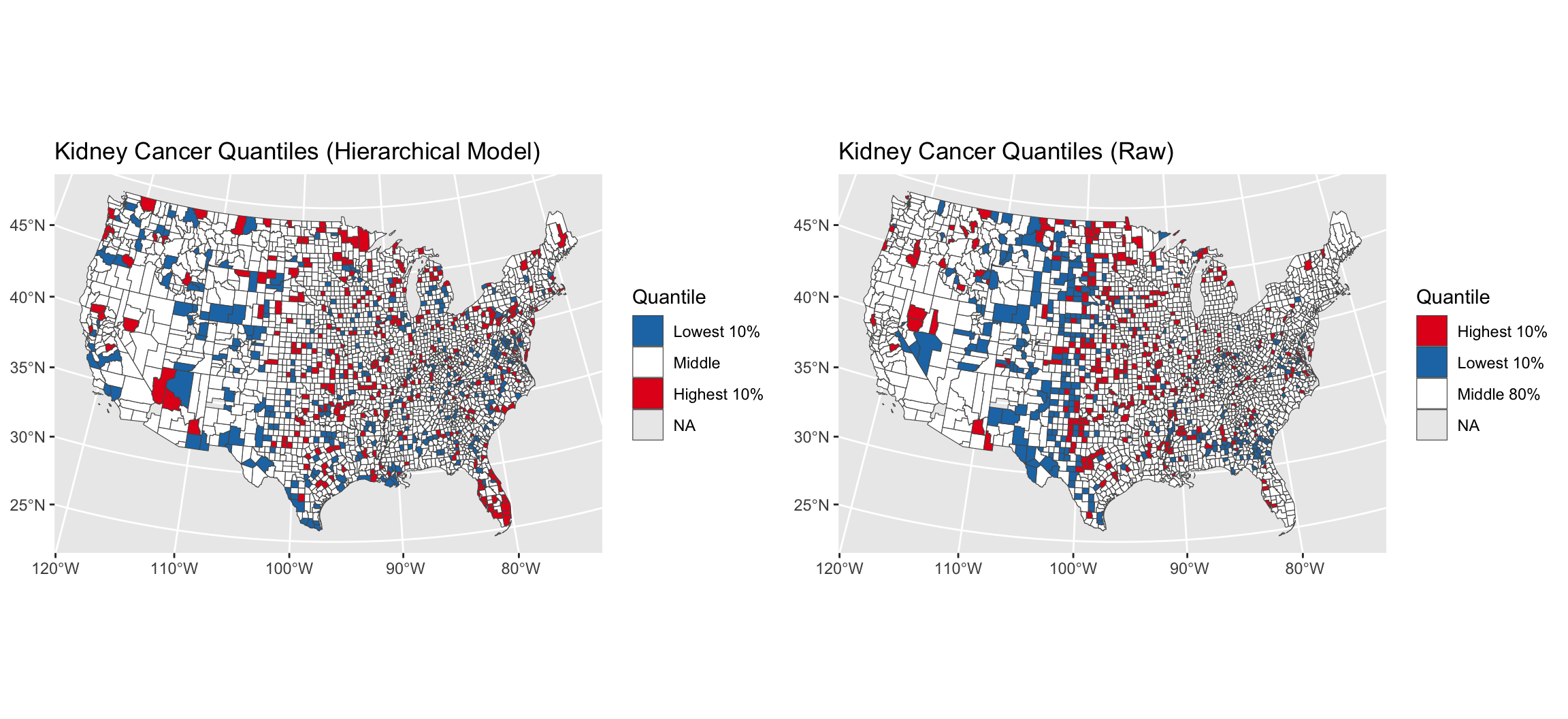
Kidney cancer death rates in the US
- Kidney cancer death rates in the US, by county
- Is there a geographic pattern? If so, why?
Data source: Gelman et al. 2004. Bayesian Data Analysis.
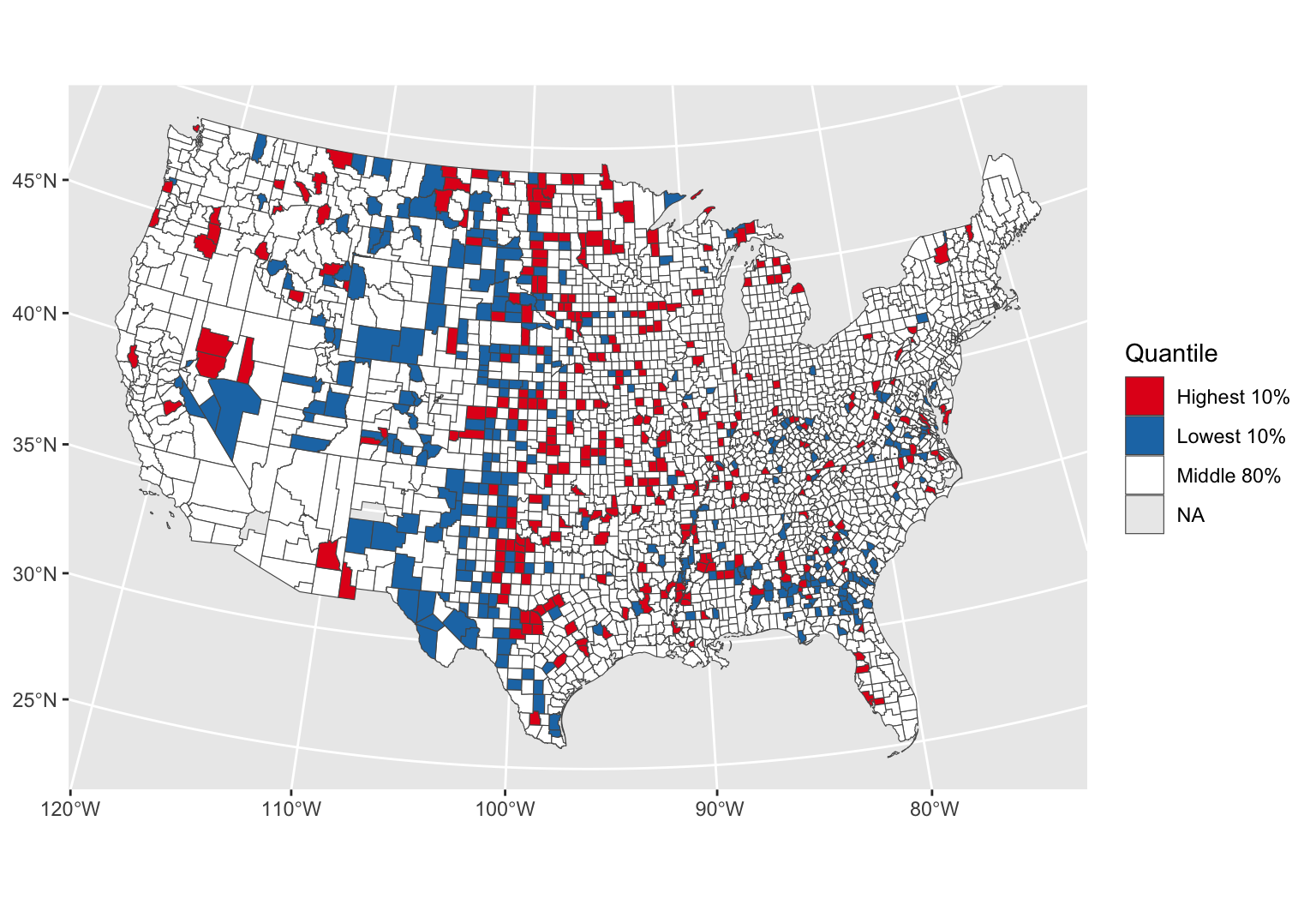
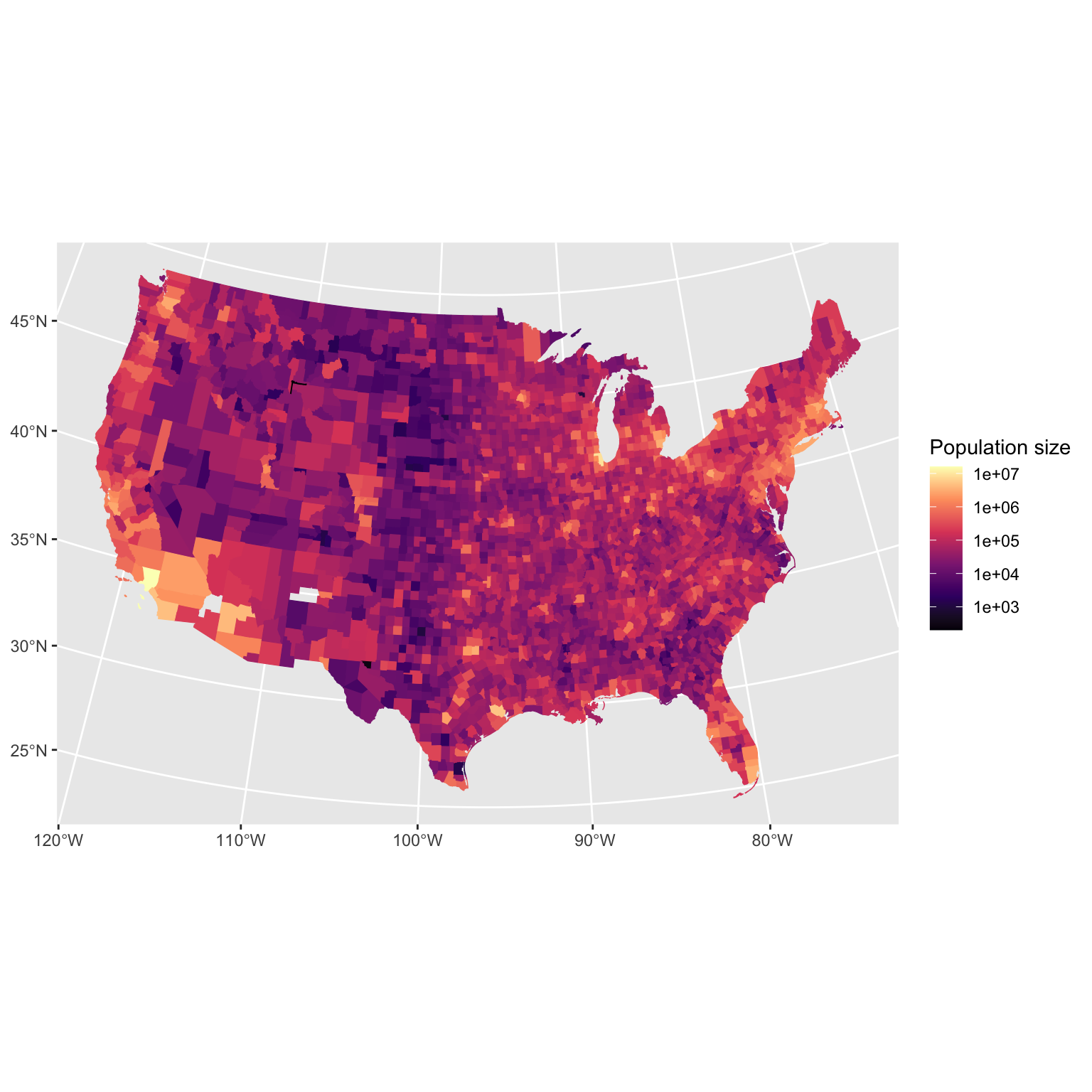
Data source: Gelman et al. 2004. Bayesian Data Analysis.
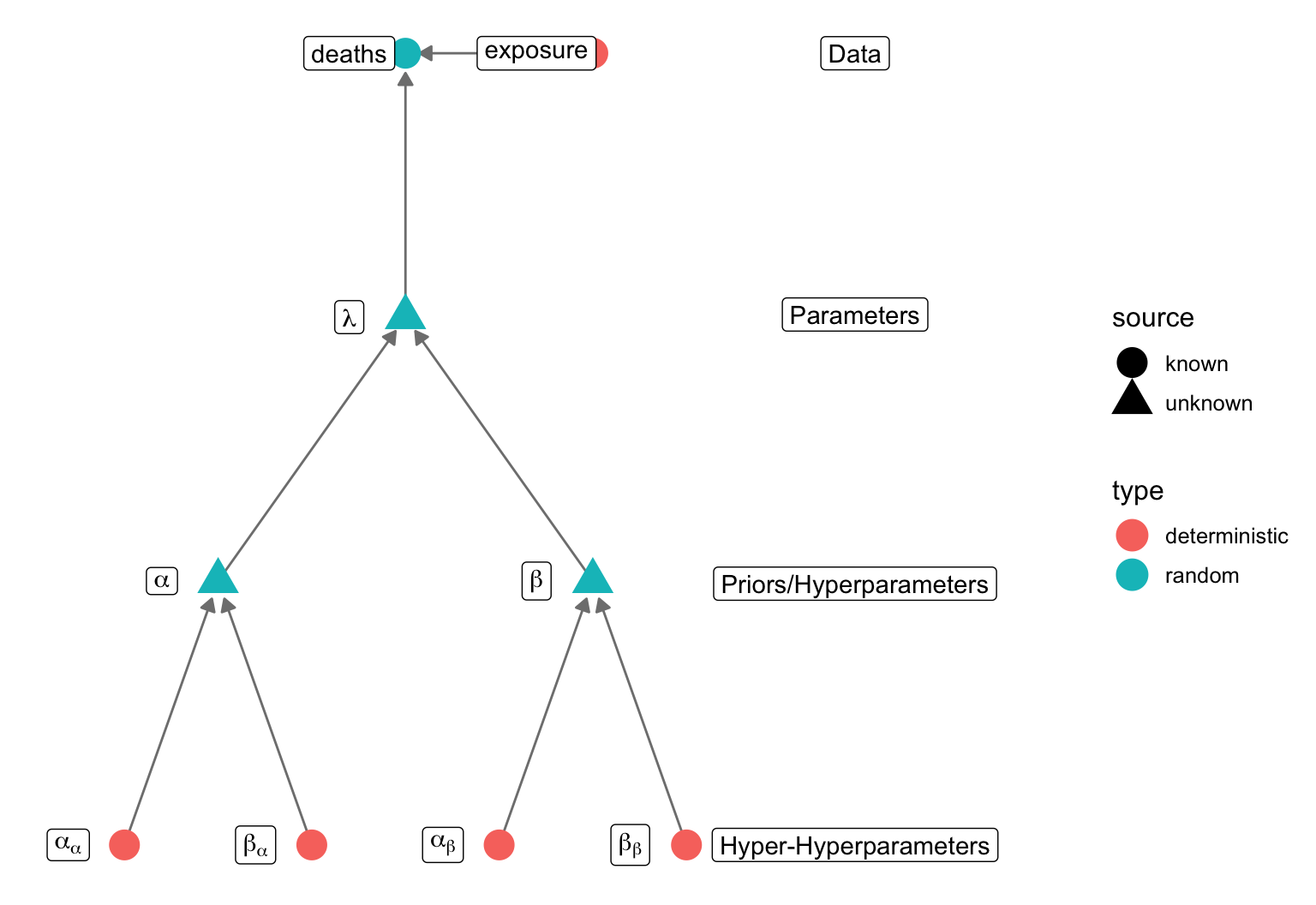
data {
int <lower = 1> n;
int <lower = 1> n_counties;
int <lower = 0> deaths [n];
int <lower = 0> population [n];
int <lower = 0, upper = n_counties> county_id [n];
// hyper-hyper parameters, for the hyperprior
real <lower = 0> a_alpha;
real <lower = 0> a_beta;
real <lower = 0> b_alpha;
real <lower = 0> b_beta;
}
transformed data {
// cancer is rare, lets make the numbers more reasonable
vector <lower = 0> [n] exposure;
for(i in 1:n)
exposure[i] = population[i] / 1000.0;
}
parameters {
vector <lower = 0> [n_counties] lambda;
// prior hyperparameters for lambda are now parameters we will estimate!
real <lower = 0> alpha;
real <lower = 0> beta;
}
model {
for(i in 1:n) {
int j = county_id[i];
deaths[i] ~ poisson(exposure[i] * lambda[j]);
}
// prior for lambda
lambda ~ gamma(alpha, beta);
// hyperpriors for alpha and beta
alpha ~ gamma(a_alpha, a_beta);
beta ~ gamma(b_alpha, b_beta);
}
generated quantities {
// save the overal mean and variance in cancer rate
real lambda_mu = alpha/beta;
real lambda_var = alpha/beta^2;
}
trees = fread("../vu_advstats_students/data/treedata.csv")
tsuga = trees[grep("Tsuga", species_name)]
# remove NAs
tsuga = tsuga[complete.cases(tsuga), ]
head(tsuga)
## n died year species_name annual_mean_temp tot_annual_pp prior_mu
## <int> <int> <int> <char> <num> <num> <num>
## 1: 5 3 1989 Tsuga canadensis 3.849333 1003.0000 0.02222166
## 2: 6 0 1989 Tsuga canadensis 3.452000 1076.1333 0.02222166
## 3: 3 0 1997 Tsuga canadensis 3.620000 1099.4000 0.03245619
## 4: 4 4 1989 Tsuga canadensis 4.596000 989.4667 0.02222166
## 5: 3 0 1994 Tsuga canadensis 4.244667 1116.2000 0.02816339
## 6: 7 0 2002 Tsuga canadensis 4.730000 1137.2667 0.04108793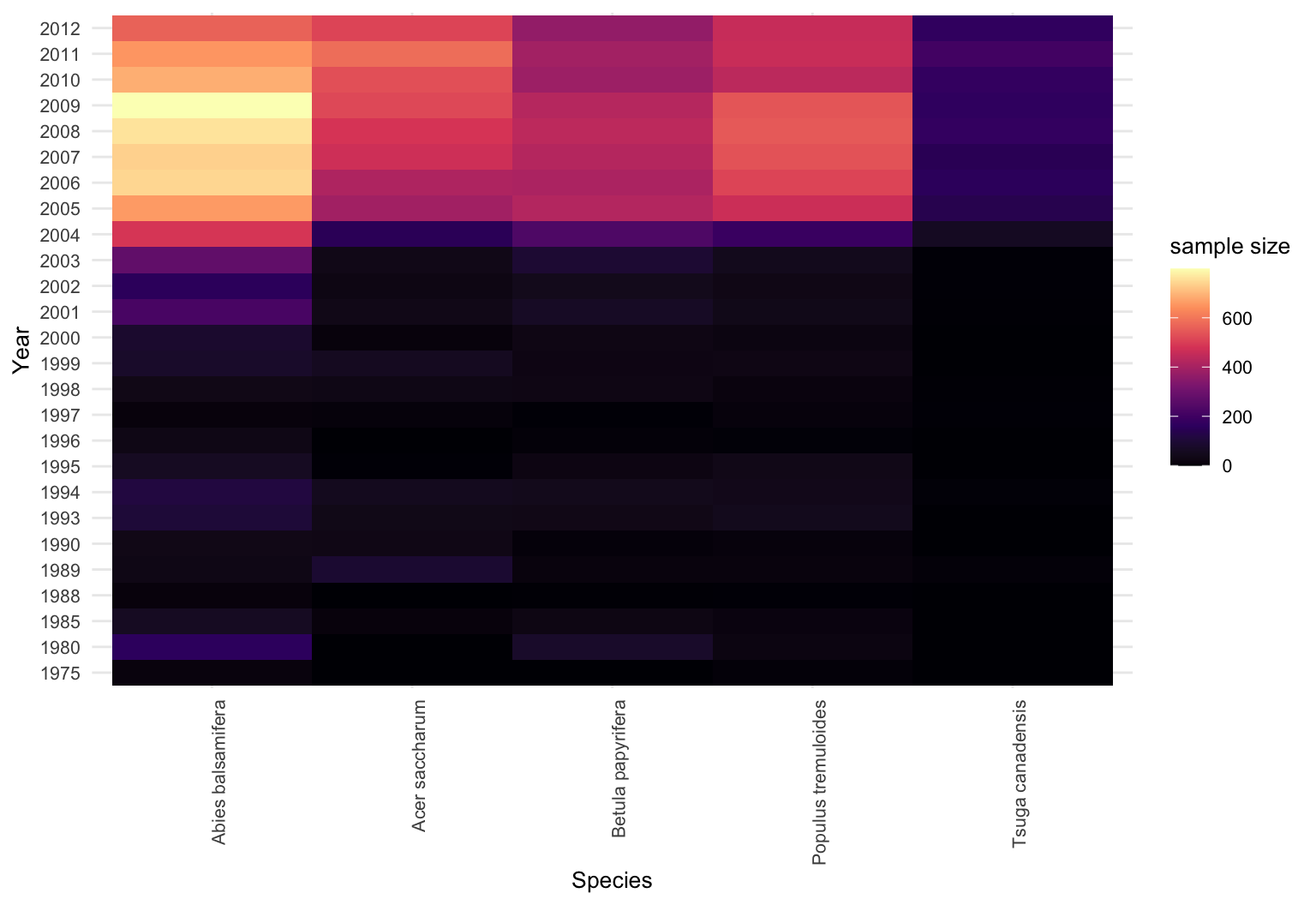
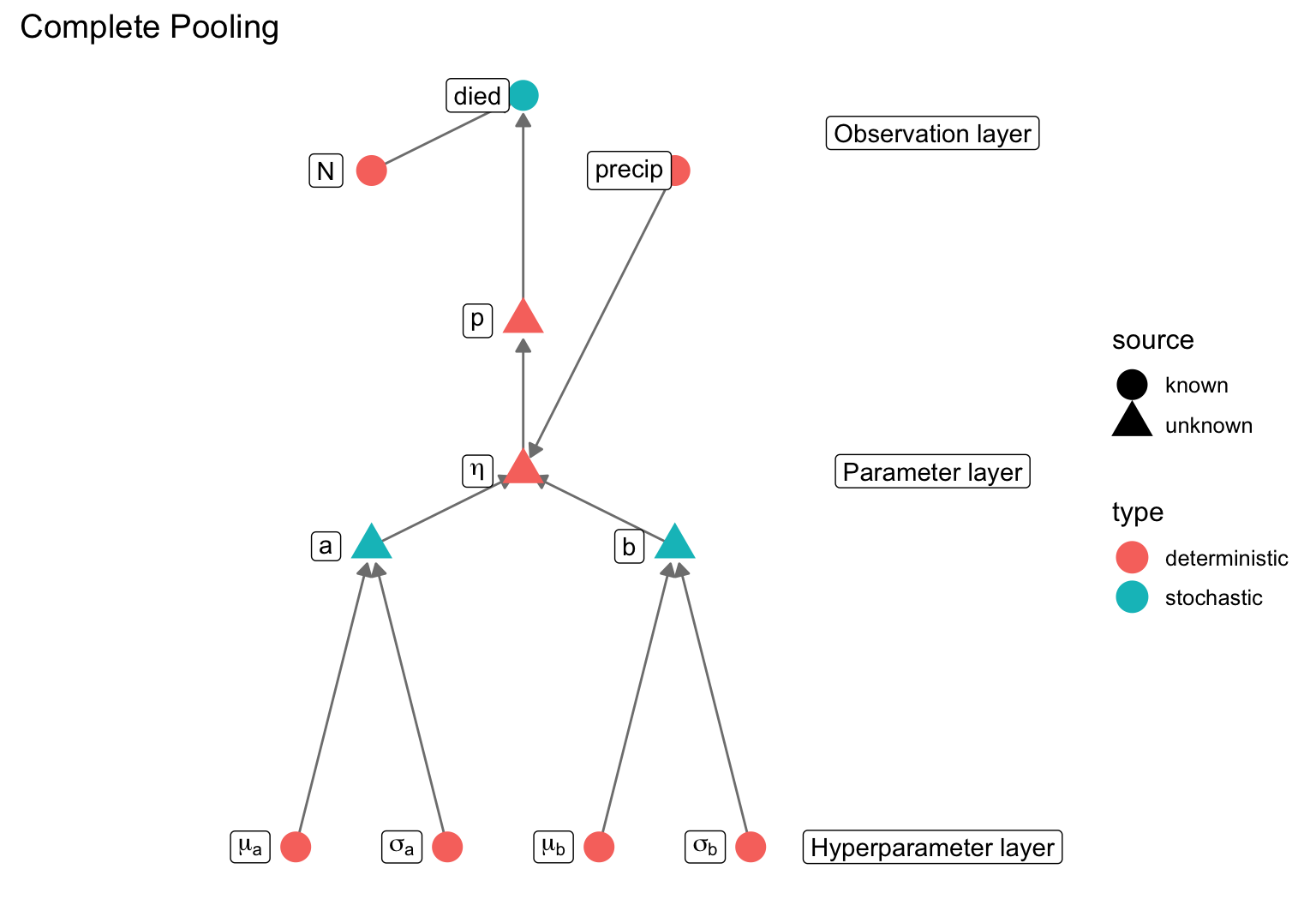
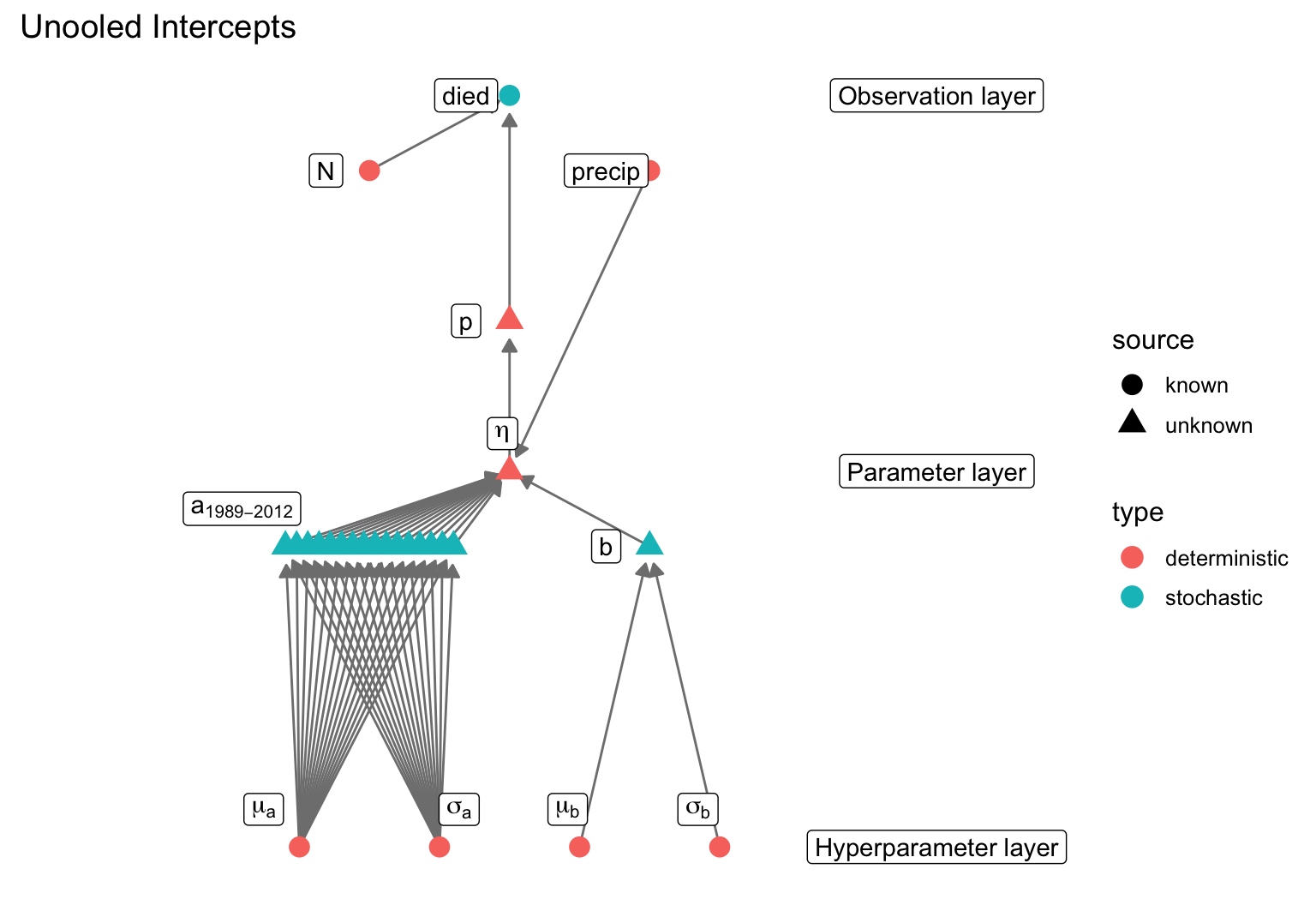
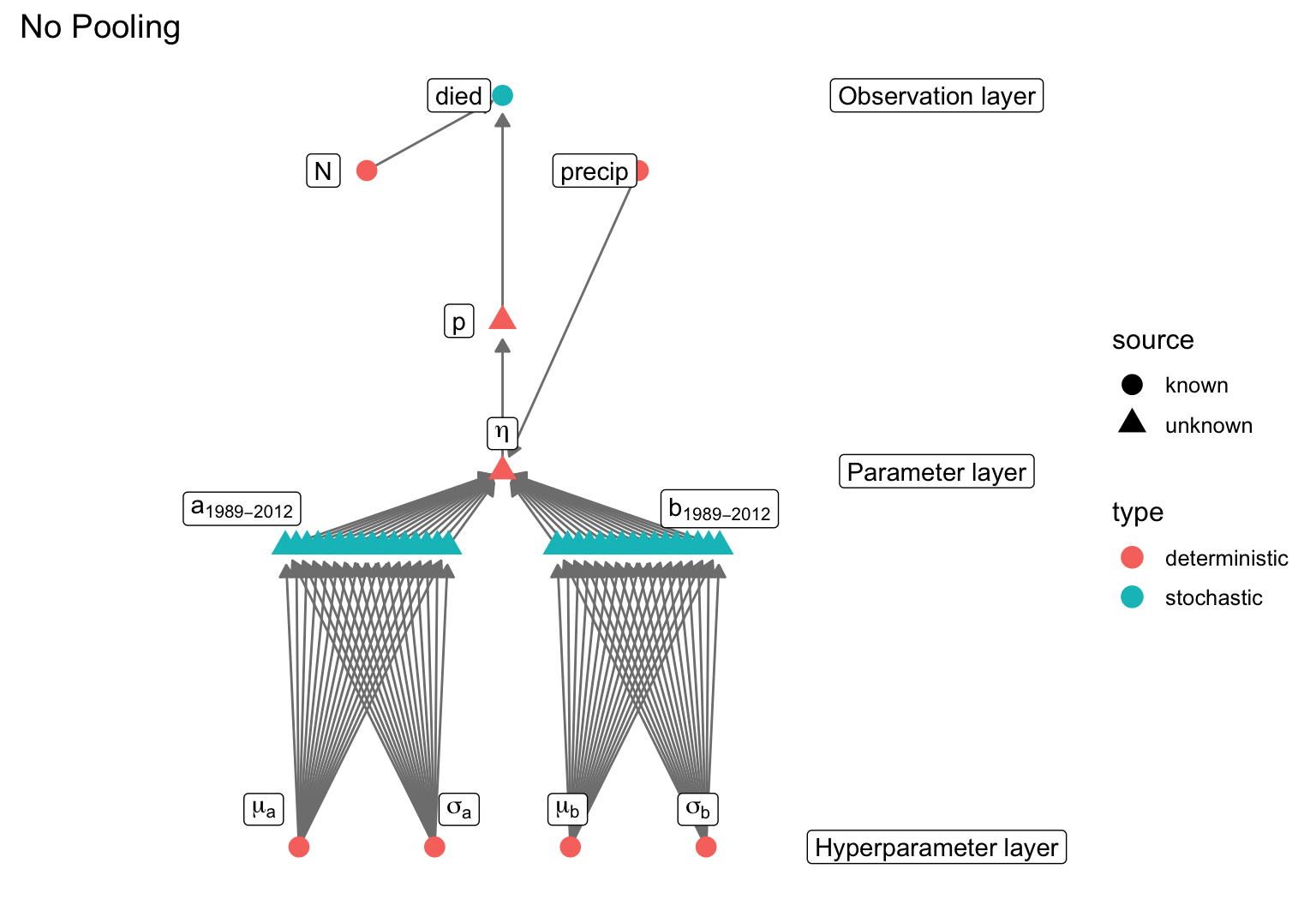
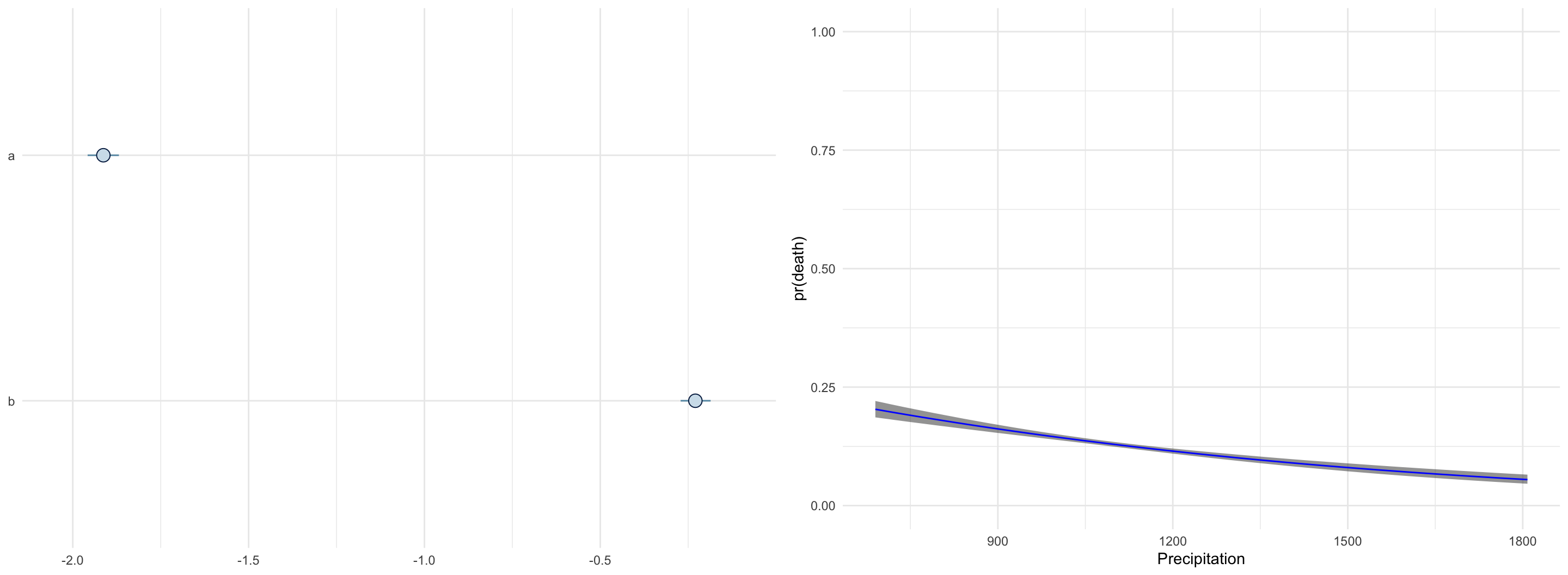
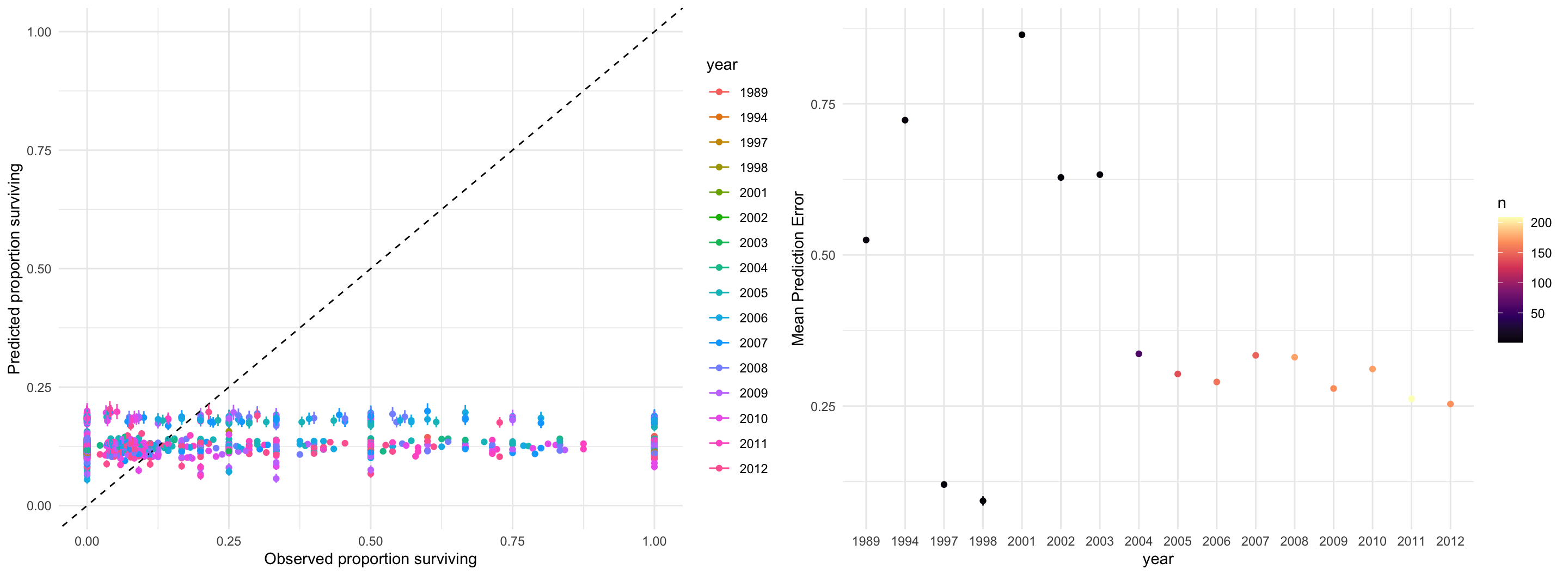
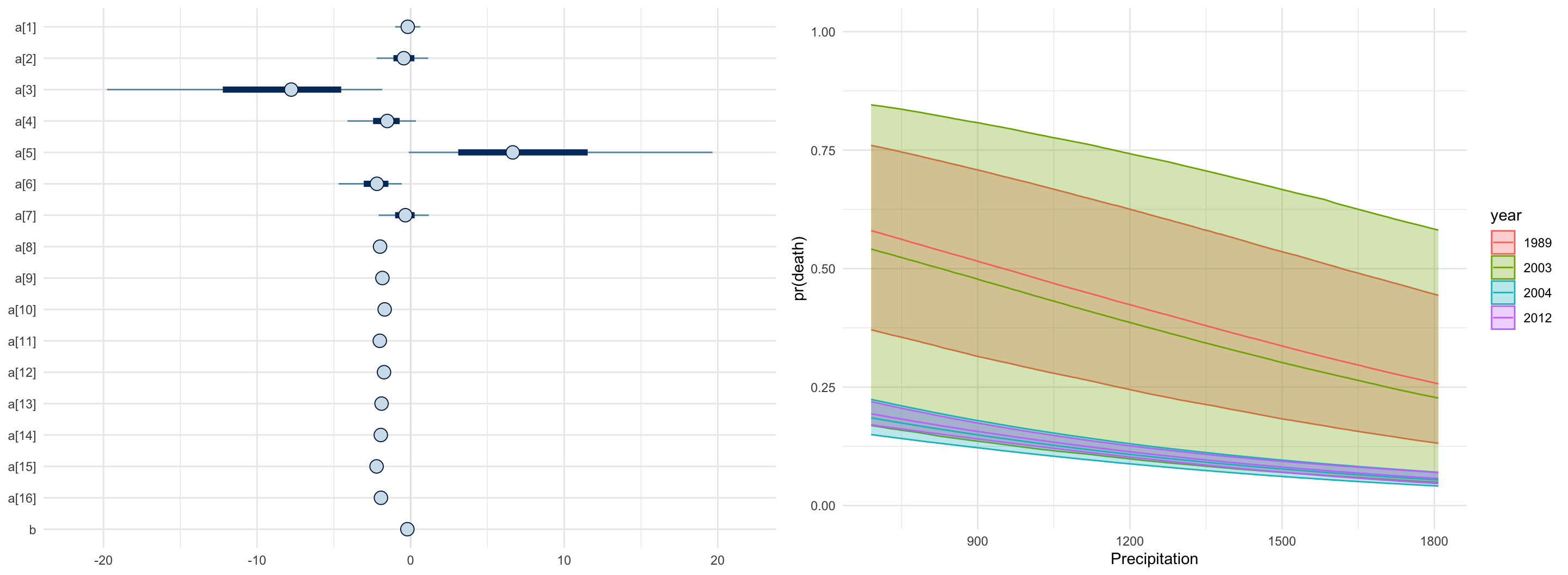
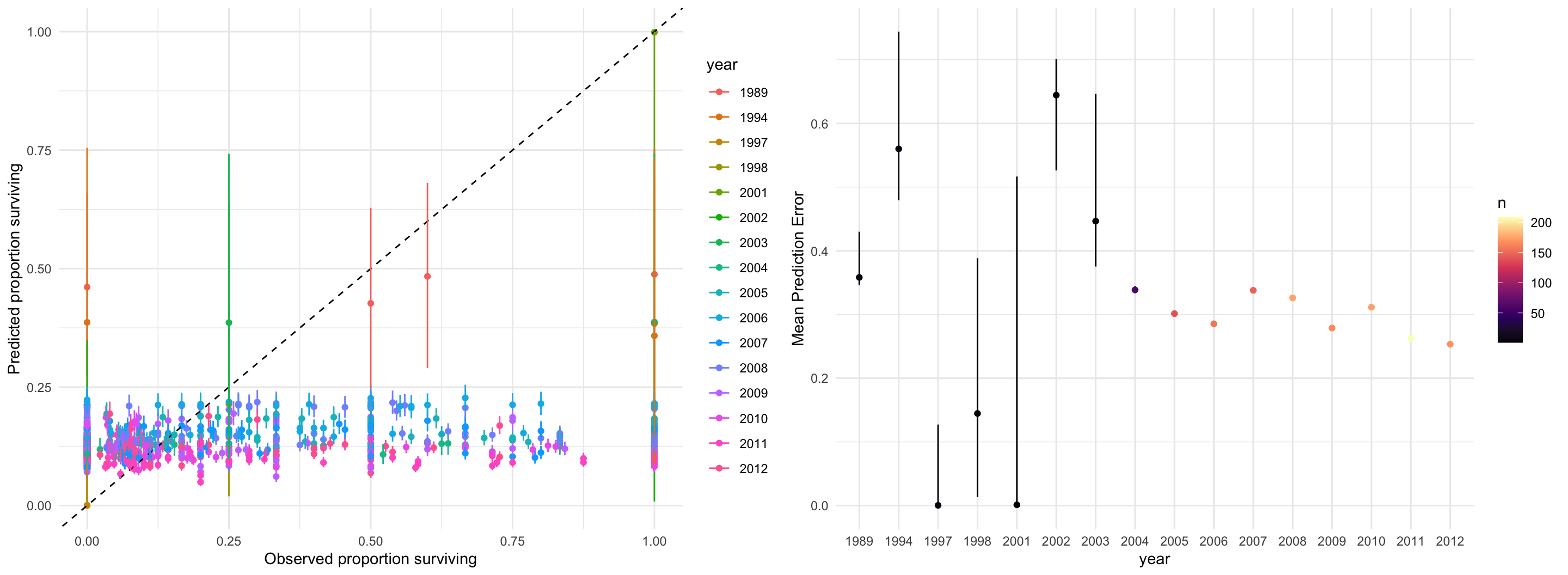
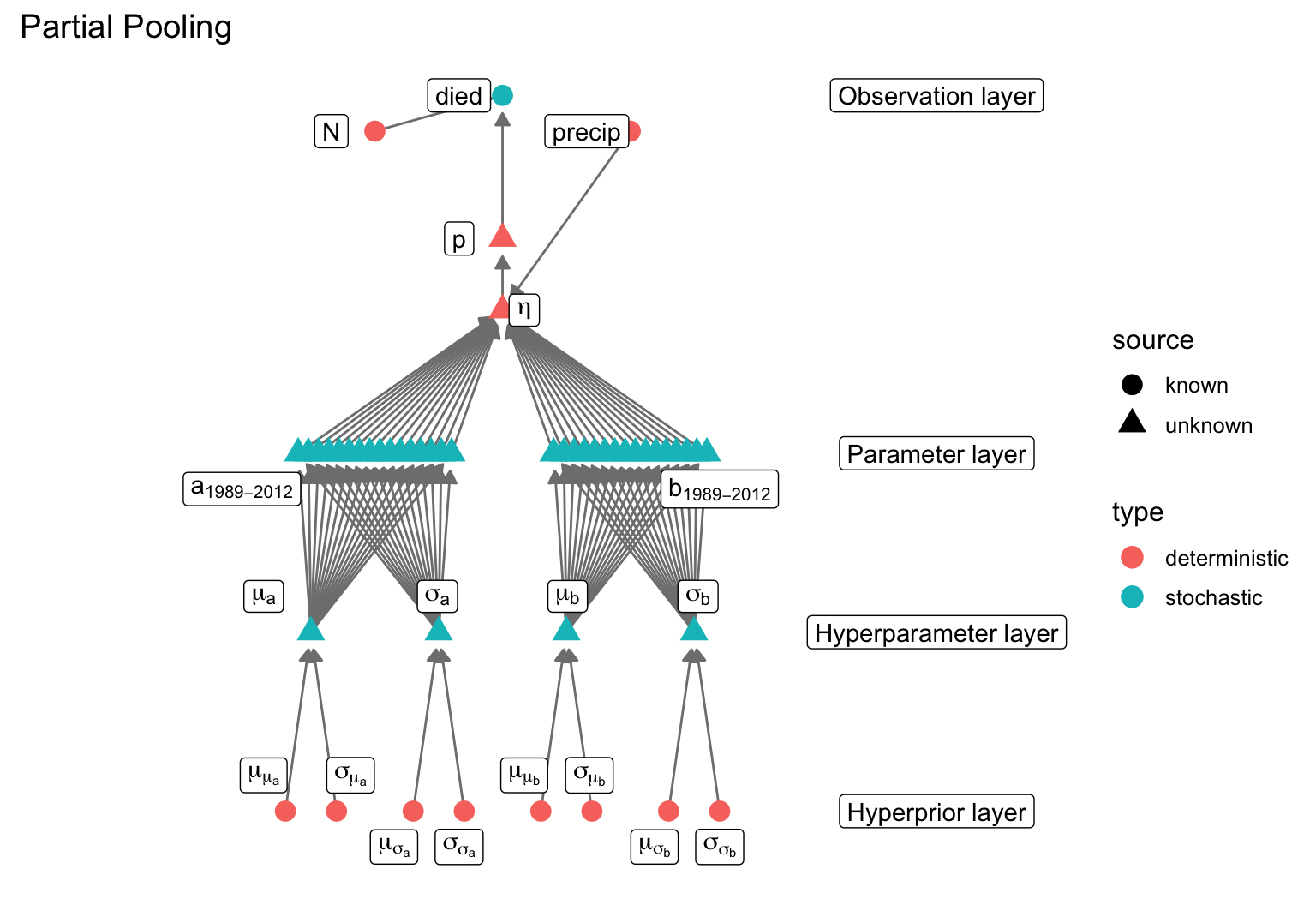
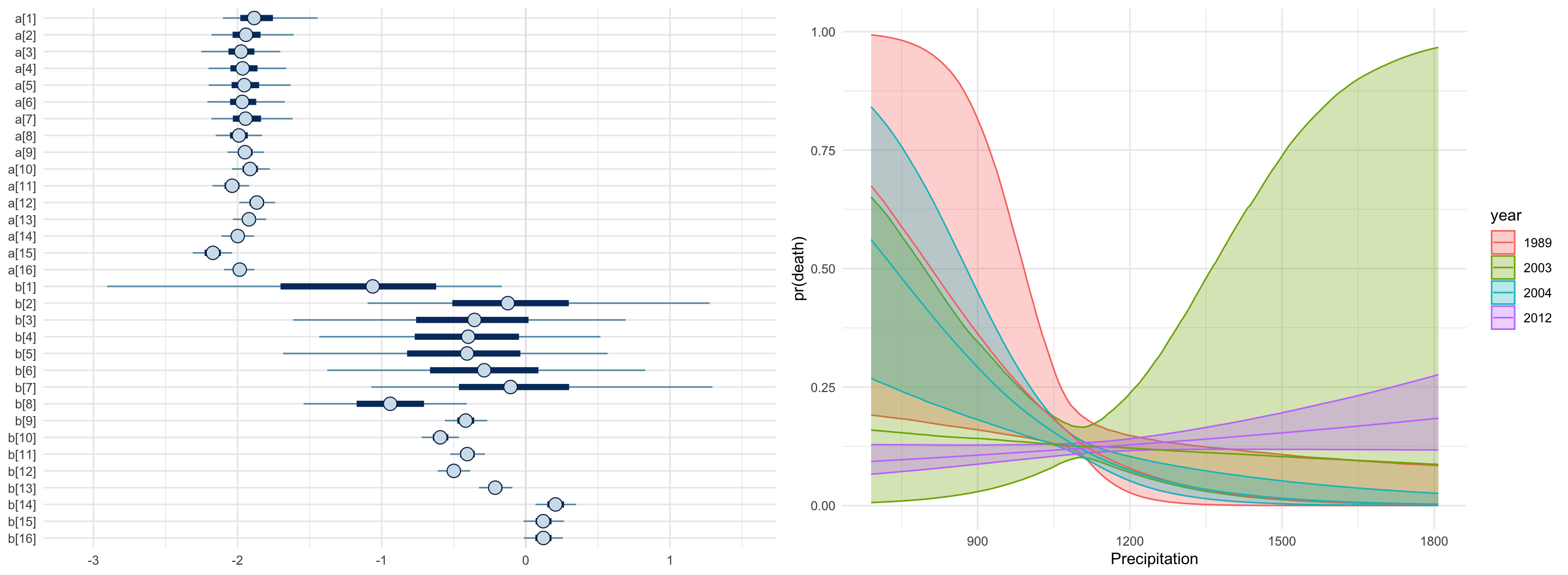
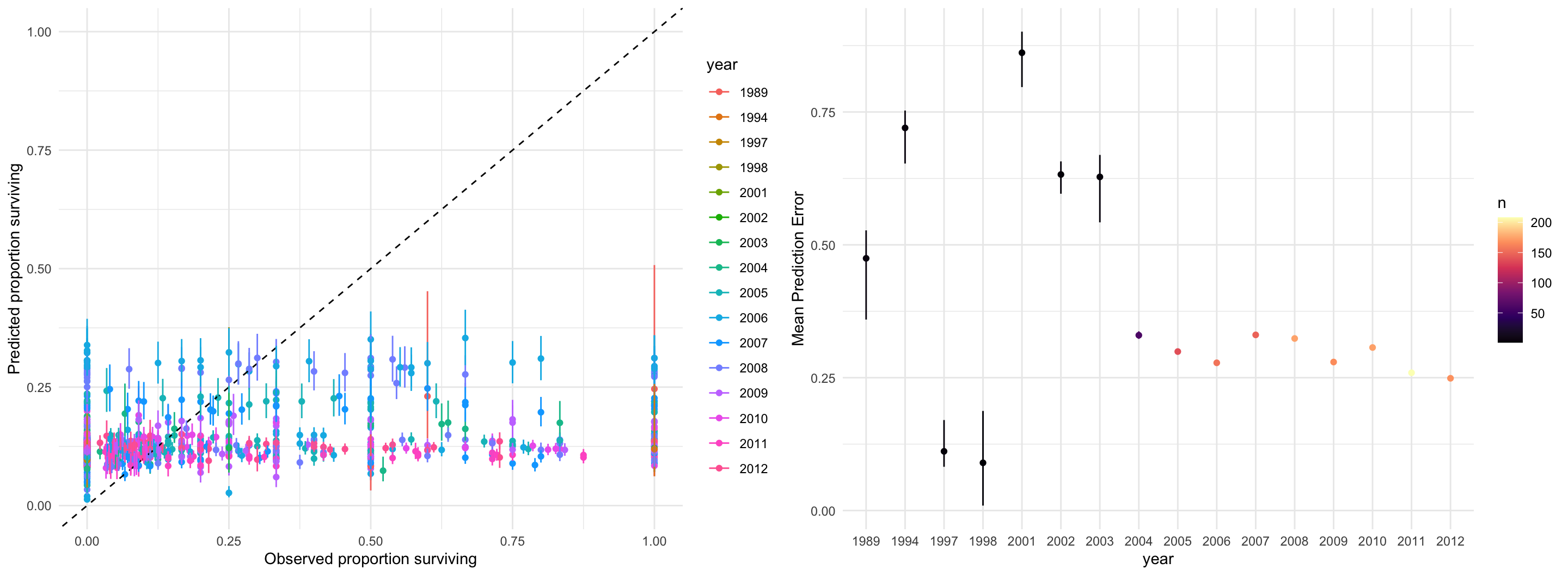
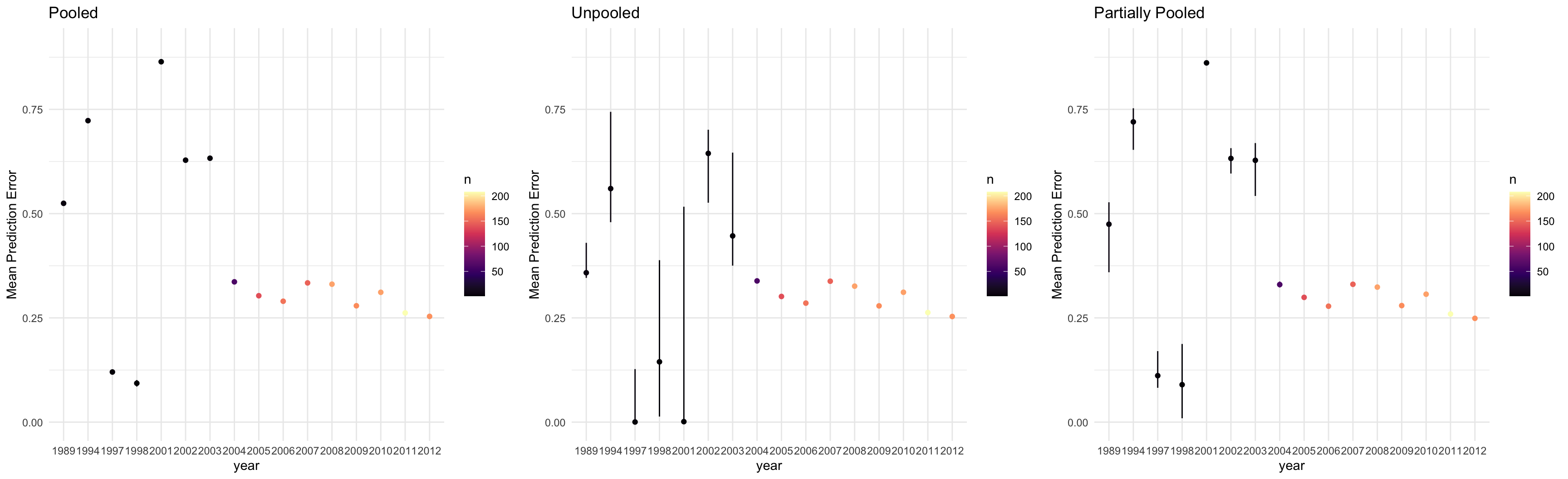
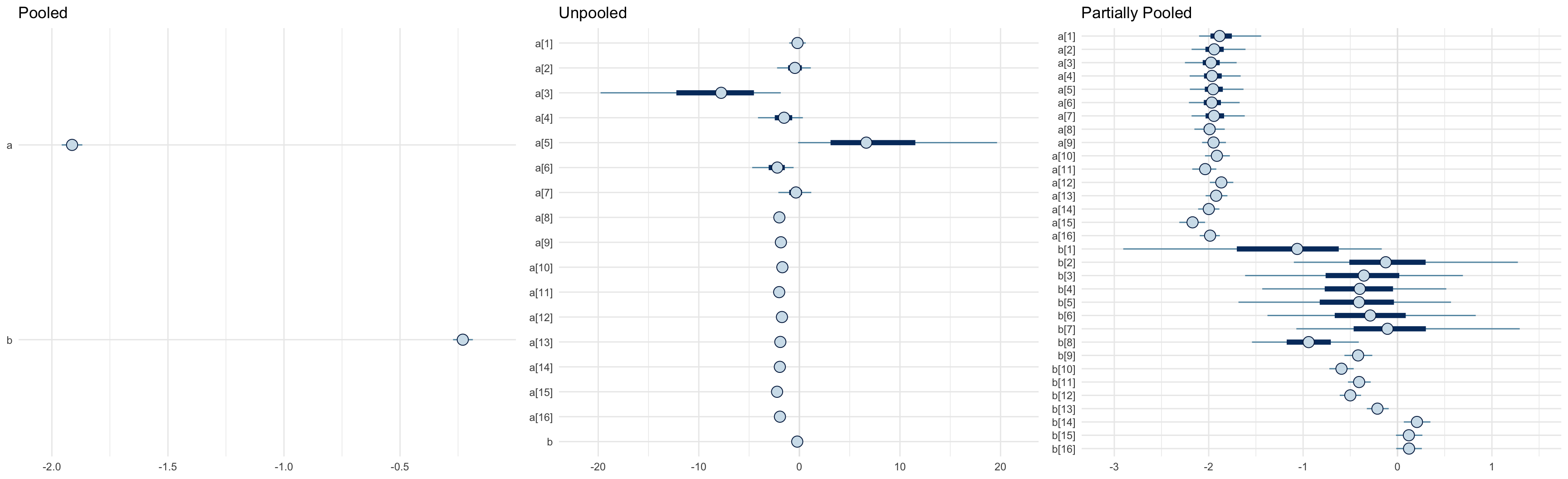
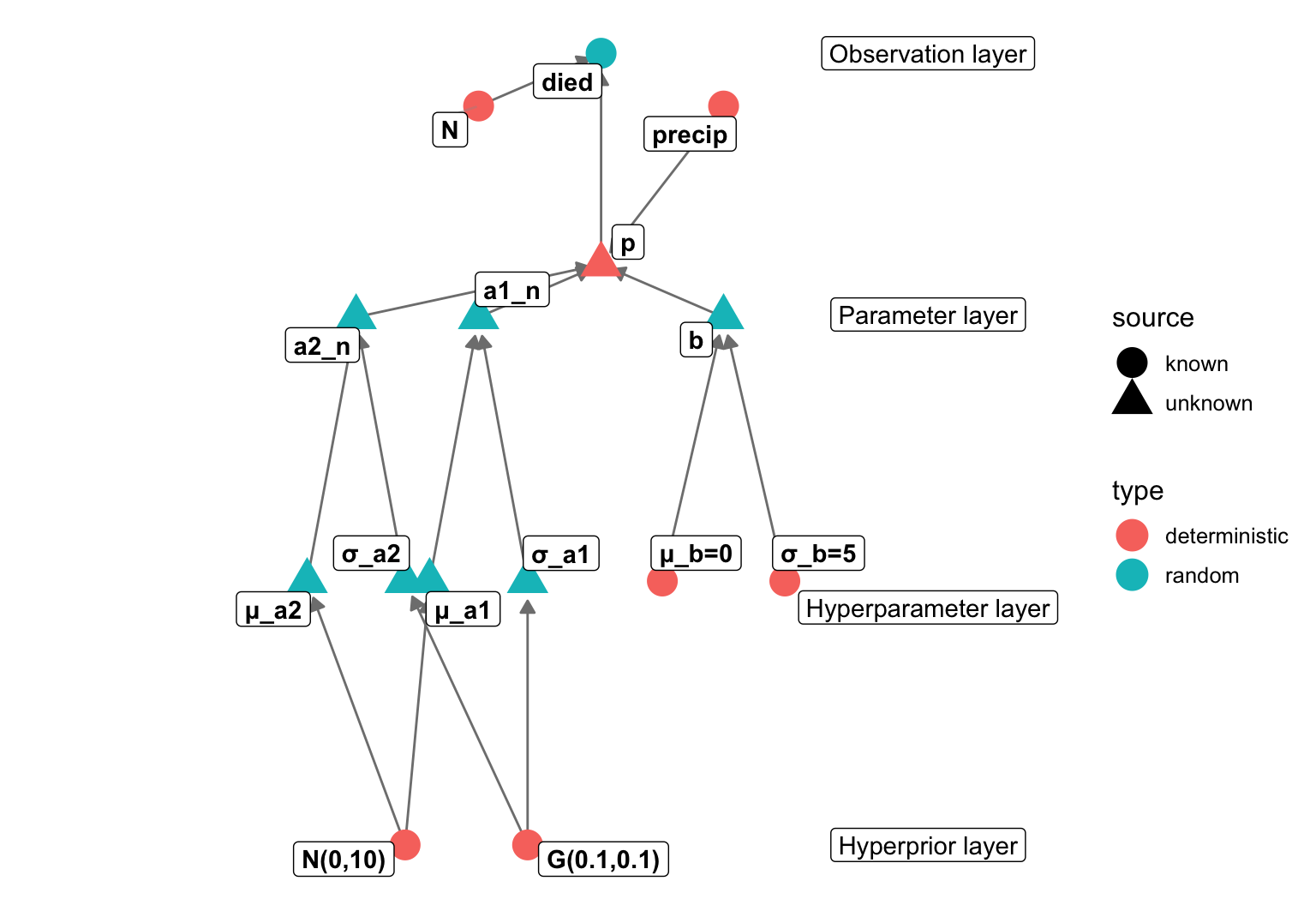
data {
int n; // number of data points
int died [n]
int N[n];
vector [n] precip;
// group-level objects
int <lower=1> n_group1;
int <lower=1, upper=n_group1> group1_id [n];
int <lower=1> n_group2;
int <lower=1, upper=n_group2> group2_id [n];
}
parameters {
vector [n_group1] a1;
vector [n_group2] a2;
// hyperparameters
real a1_mu;
real <lower=0> a1_sig;
real a2_mu;
real <lower=0> a2_sig;
}
transformed parameters {
vector [n] pr;
for(i in 1:n)
pr[i] = inv_logit(a1[group1_id[i]] + a2[group2_id[i]] + b*precip[i]);
}
model {
died ~ binomial(N, pr); // likelihood
a1 ~ normal(a1_mu, a1_sig); // hierarchical prior for a1
a2 ~ normal(a2_mu, a2_sig); // hierarchical prior for a2
// hyperpriors
a1_mu ~ normal(0,10)
a2_mu ~ normal(0,10)
a1_sig ~ gamma(0.1, 0.1);
a2_sig ~ gamma(0.1, 0.1);
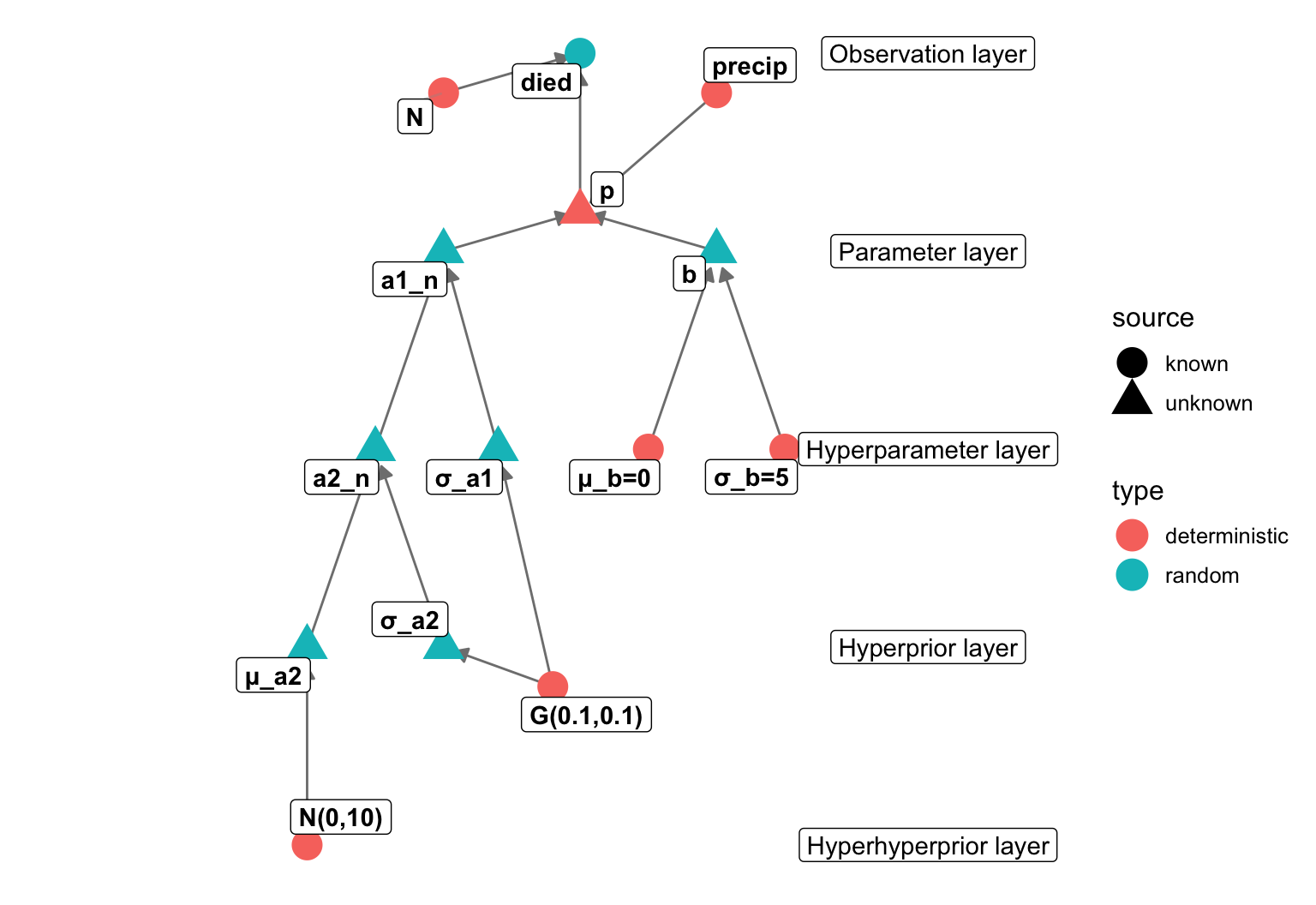
}
data {
int n; // number of data points
int died [n]
int N[n];
vector [n] temperature;
// group-level objects
int <lower=1> n_group1;
int <lower=1, upper=n_group1> group1_id [n];
int <lower=1> n_group2;
int <lower=1, upper=n_group2> group2_id [n_group1];
}
parameters {
vector [n_group1] a1;
vector [n_group2] a2;
// hyperparameters
real <lower=0> a1_sig;
real a2_mu;
real <lower=0> a2_sig;
}
transformed parameters {
vector [n] pr;
for(i in 1:n)
pr[i] = inv_logit(a1[group1_id[i]] + b*precip[i]);
}
model {
died ~ binomial(N, pr); // likelihood
for(i in n_group1)
a1 ~ normal(a2[i], a1_sig); // hierarchical prior for a1
// hyperpriors
a2 ~ normal(a2_mu, a2_sig); // hierarchical prior for a2
a1_sig ~ gamma(0.1, 0.1);
// hyperhyperprior
a2_mu ~ normal(0,10)
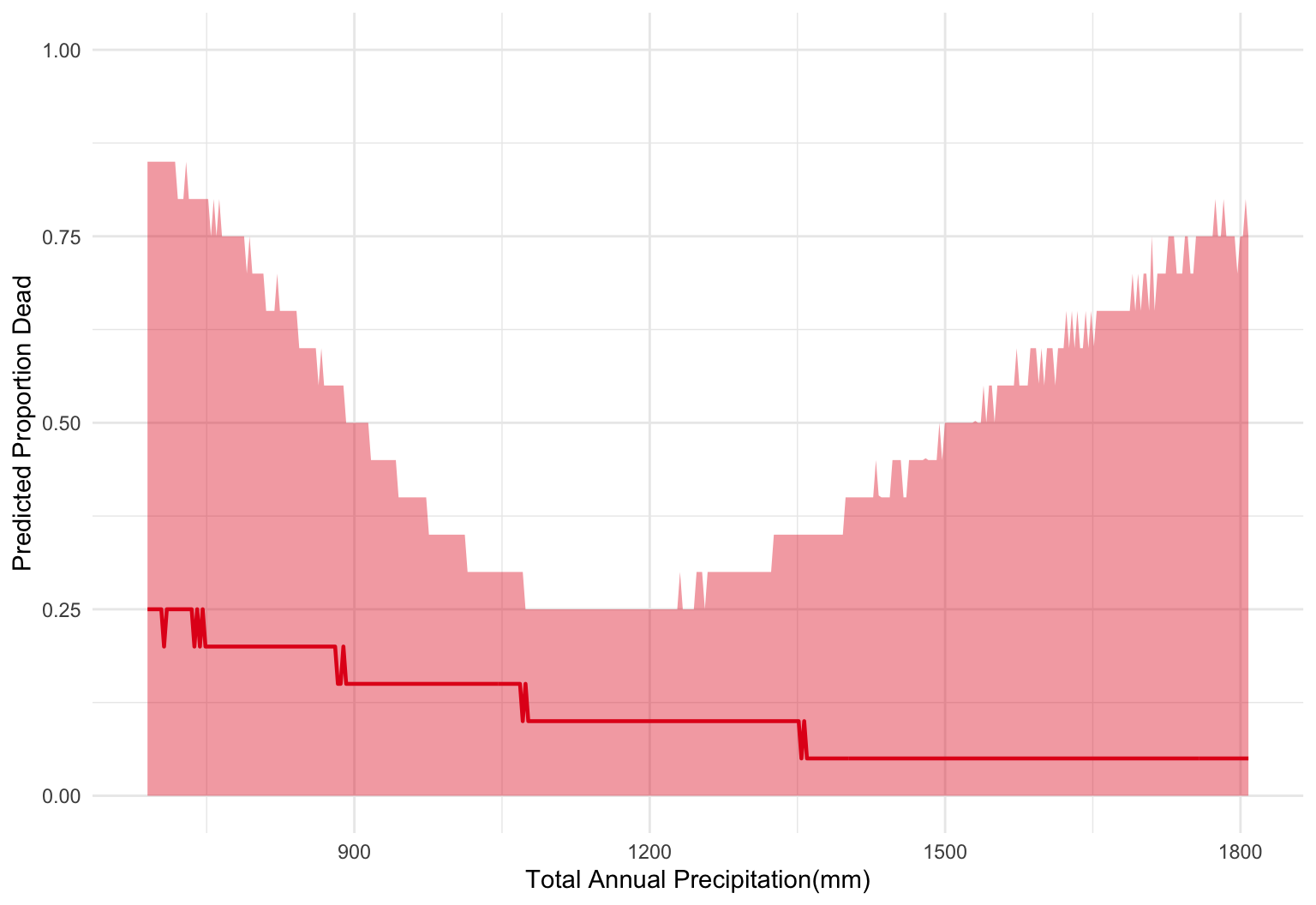
}newx = seq(min(standat$precip), max(standat$precip), length.out=400)
pars = data.frame(as.matrix(fit_ppool, pars=c("a_mu", "a_sig", "b_mu", "b_sig")))
# For our hypothetical, we need to decide how many trees we would see
# more trees means less sampling uncertainty
N = 20
sims = mapply(sim1, amu = pars$a_mu, asig = pars$a_sig,
bmu = pars$b_mu, bsig = pars$b_sig,
MoreArgs = list(N = 20, precip = newx))
sim_quantiles = apply(sims, 1, quantile, c(0.5, 0.05, 0.95))